Mucomyst Contrast Prophylaxis ProtocolThe following is the standard Mucomyst prophylaxis protocol used in some patients with chronic renal insufficiency and in whom an intra-venous or intra-arterial contrast exam is required. The referring physician, who may wish to consult with an RMG physician, must prescribe Mucomyst. After the contrast exam is complete, the patient should be hydrated (oral or intravenous) and should have renal function follow-up by the referring physician.
The pre-treatment protocol (Reference: NEJM 2000; 343: 180-184) is as follows:
Mucomyst (Acetylcysteine) solution 600 mg orally twice a day for four (4) doses beginning the day before the contrast exam.
Mucomyst (Acetylcysteine) solution comes in 10% (100mg/ml) and 20% (200mg/ml) concentrations in 4, 15, or 30 ml vials. Most pharmacists dispense the doses in individual dose syringes and have patients squirt each dose into a glass of soda.
Feb 15, 2011
Traumatic LP-Bloody tap wbc and pro. adjustment
Bloody tap if: 1 WBC/500-1,000 RBC assuming the hematocrit is normal or 1 mg/dL protein increase/1,000 RBC
Jun 15, 2008
Jan 25, 2008
Mar 30, 2007
HEREDITARY THROMBOPHILIA
Remember that the most common reason is the acquired forms (Like Antiphospholipid antibody ) and not the hereditary forms ।
Testing for hereditary causes :
1-PT and aPTT(the first to start)
2-AT deficiency: Antithrombin deficiency(heparin cofactor 1)। inhibits thrombin and fact x॥ also heparin needs this factor to work !! , when can’t reach therapeutic PTT on heparin drip !! 2 type। don’t waste your time with heparin
3-Protein C deficiency: check activity first .2 type ,pro c and Z are Vit K dependent
4- Protein S deficiency: check activity first .2 type ,it binds to C4B with goes up in acute phase and will drop protein s level, you cab check c4b or free pro S
5-APC Resistance :activated Pro C resistance nothing to do with Pro C ! inhibits fac V and VIII, if positive check Factor V Leiden
6- Factor V Leiden: most common , major cause of APCR
7- Prothrombin G20210A Mutation :mutation will cause excess of prothrombin(II)
8-Factor VIII Excess (check fibrinogen at then same time, it is a acute phase pro and ..) excess level of 800 %
9-Homocysteine, excess level , treatment is not anticoagulation B12,6, folate
-MTHFR
10-Factor XII deficiency :even though it’s a part of intrinsic factor and one might think should cause bleeding it does the opposite (also involve in converting plasminogen to plasmin)
11-Protein Z deficiency: Vit K dependent like pro
12-Heparin cofactor II deficiency :inhibits thrombin (not factor X)
13-TFPI: tissue factor pathway inhibitor deficiency;inhibits X and VII
14-PAI-I :plasminogen activator inhibitor -1 ,inhibits fibrinolysis. Expensive, test if everything else negative.
15-Elevated factor XI and V and II
Balance between adequate blood flow in normal vessels and rapid cessation of bleeding in injured vesselsInteraction between coagulation cascade and fibrinolysisBegins with endothelial injury à platelet activation à activation of clotting cascade à anticoagulants à fibrinolysis à resolution of clotImbalance in HemostasisThrombosis:Multigenic/MultifactorialTwo hit hypothesis: more than one risk factor required to develop thrombosisAntithrombin Antithrombin Manufactured in liverOther designationsHeparin cofactor I Antithrombin IIISerine protease inhibitorRequires heparan or heparin to functionHeparin requires antithrombin to functionAntithrombin Natural anticoagulant activityInactivates thrombin and factors Xa, IXa and XIaForms 1:1 complex with themDeficiency results in thrombosisVenous – more commonArterial Antithrombin DeficiencyEstimated frequency = 1%Types of deficiencyType I - quantitativeA = normal molecule, decreased amountB = abnormal molecule, abnormal amountType II – qualitative, abnormal moleculeA = activity and heparin binding siteB = activity onlyC = binding site onlyAntithrombin DeficiencyLaboratory testingAntithrombin activity or functionalChromogenic, if availableTreatmentAnticoagulationAntithrombin III concentrate (Thrombate III)Activation of Protein CProtein CManufactured in liverVitamin K dependentZymogen (must be activated)Thrombin combines with thrombomodulinRequires Protein S as cofactorProtein C DeficiencyNatural anticoagulant activityInactivates factors Va and VIIIaInactivates PAI-IDeficiency results in thrombosisVenous – more commonArterialPurpura fulminansHomozygous childrenPurpura FulminansProtein C DeficiencyEstimated frequency = 5%Types of deficiencyType I – quantitativeDecreased activity & decreased amountType II – qualitativeDecreased activityNormal amountProtein C DeficiencyLaboratory testingProtein C Activity or functionalChromogenic, if availableTreatmentAnticoagulationWarfarin-induced skin necrosis may occurProtein C concentrate Warfarin-induced Skin NecrosisProtein SManufactured in liver, endothelial cells, megakaryocytes, testes, kidney & brainVitamin K dependentMost of Protein S circulates bound to C4b binding proteinAcute phase reactantIncreases with inflammationProtein SFree Protein S is active formNatural anticoagulant activityCofactor for Protein CCombined with Protein CInactivates factors Va and VIIIaAloneWeak activity to inactivate factors Va, VIIIa and XaProtein S DeficiencyDeficiency results in thrombosisVenous – common with inactivityArterial – common in cerebral vesselsPurpura fulminansEstimated frequency = 3%Protein S DeficiencyTypes of deficiencyType I – quantitativeDecreased total, free and functionalType II – qualitativeNormal total and freeDecreased functionalType III – associated with increased C4bNormal totalDecreased free and functionalProtein S DeficiencyLaboratory testingProtein S activity or functionalProtein S freeC4b binding proteinChromogenic, if availableTreatmentAnticoagulationWarfarin-induced skin necrosisFactor XII Factor XIIManufactured in liverHageman factorSerine proteaseActivated by kallikrein (Fletcher Factor)Factor XIIa linked to kinin system, intrinsic fibrinolysis & complement systemsConverts plasminogen to plasminEstimated frequency of deficiency is lowFactor XII DeficiencyDeficiency decreases fibrinolysisDeficiency results in thrombosisVenous – more commonArterial – rareLaboratory testingFactor XII levelsTreatmentAnticoagulation Protein ZProtein ZVitamin K dependentCofactor for PZ-dependent protease inhibitor (PZI)Results in inactivation of factor XaFrequency of deficiency unknownDeficiency results in venous thrombosisLaboratory testing = Protein Z levelsTreatment = anticoagulationHeparin Cofactor IIHeparin Cofactor IIGlycoprotein, serine protease inhibitor familyActivated by heparan, heparin or dermatan sulfateResults in inhibition of thrombinNO effect on factor XaHeparin Cofactor II DeficiencyDecreased inhibition of thrombinDeficiency results in venous thrombosisActual risk is unknown at this timeLaboratory testingHeparin cofactor II levelsTreatmentAnticoagulation Tissue Factor Pathway Inhibitor (TFPI)TFPILocated in endothelial cellsBound to heparan sulfateHeparin increases plasma concentrationCombines with free factor Xa à Xa:TFPIXa:TFPI combines with VIIa:TFDecreased production of factor XInactivation of VIIa:TFTFPI DeficiencyFrequency unknownDeficiency causes:Venous thrombosisIntrauterine lethality (fetal loss)Neointimal proliferationLaboratory testingTFPI levelsTreatmentAnticoagulation Activated Protein C Resistance(APCR)APC ResistanceFrequency difficult to determine – may be as high as 30-50%Normal Protein C and Protein SNormal levels of activated Protein C with normal activityAPC unable to combine with factor Va for inactivationAPC ResistanceDefective factor VaReceptor for APC defectiveAPC unable to bind & inactivateNow looking at areas on factor VIIIaDefects in APC binding siteResults in venous & arterial thrombosisLaboratory testing = APC resistance assayTreatment = anticoagulation Inactivation of Factor VaFactor V LeidenMajor cause of APC ResistanceFrequency estimated at 20%Single point mutation (1691 Gà A) results in 506 Arg/Gln substitutionAffects first of three APC cleavage sitesAPC cannot down-regulate Factor VaFactor Va forms prothrombinase complex with factor Xa à conversion of prothrombin to thrombinProlonged procoagulant activityFactor V LeidenMost common genetic risk factor for thrombosisVenous and arterial Heterozygotes: 3 – 10X increased riskHomozygotes: 80X increased riskLaboratory testingRT-PCR for Factor V LeidenTreatment = anticoagulationOther Factor V MutationsFactor V Hong KongSingle point mutation results in 306 Arg/GlyRare cause of venous thrombosisFactor V CambridgeSingle point mutation results in 306 Arg/ThrRare cause of thrombosisFactor V LiverpoolSingle point mutation at Arg 679Rare cause of venous thrombosisHR2 HaplotypeSeveral mutationsRisk of venous thrombosis unknown at this timeThrombin: Central Role in HemostasisProthrombin G20210A MutationProthrombin G20210AEstimated frequency is 6%Mutation in location of 3’-UT regionResults in increased levels of ProthrombinVenous and possible arterial thrombosisHeterozygotes 3X increased risk of venous thrombosis Prothrombin G20210ALaboratory testingRT-PCR testing for gene mutationDo NOT measure factor II levelsTreatmentAnticoagulation Metabolism of HomocysteineHomocysteineTwo enzymesCystathionine B-synthaseConverts homocysteine to cystathionineDefect results in homocysteinuriaMTHFR (methylene tetrahydrofolate reductase)Converts homocysteine to methionineDefect causes homocysteinemia in presence of folate depletionThree vitamins – folate, B6 & B12HyperhomocysteinemiaElevated homocysteineDamage to endothelial cellsIncreased smooth muscle cell proliferationIncreased binding of Lp(a)Decreased vascular reactivity (decreased nitric oxide synthesis) Premature atherosclerosisIncreased tissue factor & factor Va releasedDecreased thrombomodulin (protein C activation) & heparan activityBlocked endothelial cell mediated fibrinolysis Venous/arterial thrombosisPregnancy-related vascular disorders = fetal lossHyperhomocysteinemiaLaboratory testingHomocysteine levelsMTHFR mutation by RT-PCRTreatmentVitamin supplementationFolate, B6 & B12Anticoagulation, if necessaryElevated Factor VIIIEstimated frequency is 16%Manufactured in endothelial cells & megakaryocytesAcute phase reactant with levels to ~400%Factors VIIIa & IXa form tenase complex (converts factor X to Xa)Factor Xa converts factor II to thrombinElevated Factor VIIILevels >400% are dangerousResult in increased thrombinIncrease in all factor VIII componentsVenous thrombosis occursPossible arterialLaboratory testing = Factor VIII levelsTreatment = anticoagulationOther Elevated Factor LevelsRisk factor for venous thrombosisLevels above the 90th percentileInclude factors XI, V and IIProbable genetic associationTestingSpecific factor levelTreatment = anticoagulationIncreased PAI-IPlasminogen Activator Inhibitor-IInhibits fibrinolysis (conversion of plasminogen to plasmin)Decreased fibrinolysis à increased and persistent clotting (venous thrombosis)Rare but may be cause when other tests are negativeTreatment = anticoagulationLABORATORY TESTINGWhen To TestHistory of recurrent thrombosisVTE prior to 45 years of ageUnprovoked VTE – any ageThrombosis in unusual sitesThrombosis + positive family history of thrombosisVTE secondary to pregnancy, OCT, hormone replacement therapyRecurrent fetal lossEarly onset atherosclerosisLaboratory TestingPattern the testing to the clinical presentationBe sure patient is clotting (not bleeding) before ordering tests for ThrombophiliaTry to test when patient is not acutely ill and not on anticoagulantsSeveral weeks after thrombotic eventSeveral weeks after anticoagulants discontinuedRecommended Lab TestingPT & aPTTNatural anticoagulants Antithrombin activityProtein C functionalProtein S functional & freeAPC resistanceFactor V LeidenProthrombin G20210AFactor VIIIFibrinogen Additional TestingHomocysteine levelMTHFROther factor levels (for increase)FibrinolysisPlasminogenPAI-IRecurrent Fetal LossAntiphospholipid SyndromeProtein C, S & ATIIIFactor V LeidenProthrombin G20210ACombinationPoints to RememberAvoid testing during acute eventUse functional assays firstConsider age & genderAlways confirm with repeat testingIf one abnormality identified, continue testing for others (combined common)Use PCR when available
Testing for hereditary causes :
1-PT and aPTT(the first to start)
2-AT deficiency: Antithrombin deficiency(heparin cofactor 1)। inhibits thrombin and fact x॥ also heparin needs this factor to work !! , when can’t reach therapeutic PTT on heparin drip !! 2 type। don’t waste your time with heparin
3-Protein C deficiency: check activity first .2 type ,pro c and Z are Vit K dependent
4- Protein S deficiency: check activity first .2 type ,it binds to C4B with goes up in acute phase and will drop protein s level, you cab check c4b or free pro S
5-APC Resistance :activated Pro C resistance nothing to do with Pro C ! inhibits fac V and VIII, if positive check Factor V Leiden
6- Factor V Leiden: most common , major cause of APCR
7- Prothrombin G20210A Mutation :mutation will cause excess of prothrombin(II)
8-Factor VIII Excess (check fibrinogen at then same time, it is a acute phase pro and ..) excess level of 800 %
9-Homocysteine, excess level , treatment is not anticoagulation B12,6, folate
-MTHFR
10-Factor XII deficiency :even though it’s a part of intrinsic factor and one might think should cause bleeding it does the opposite (also involve in converting plasminogen to plasmin)
11-Protein Z deficiency: Vit K dependent like pro
12-Heparin cofactor II deficiency :inhibits thrombin (not factor X)
13-TFPI: tissue factor pathway inhibitor deficiency;inhibits X and VII
14-PAI-I :plasminogen activator inhibitor -1 ,inhibits fibrinolysis. Expensive, test if everything else negative.
15-Elevated factor XI and V and II
Balance between adequate blood flow in normal vessels and rapid cessation of bleeding in injured vesselsInteraction between coagulation cascade and fibrinolysisBegins with endothelial injury à platelet activation à activation of clotting cascade à anticoagulants à fibrinolysis à resolution of clotImbalance in HemostasisThrombosis:Multigenic/MultifactorialTwo hit hypothesis: more than one risk factor required to develop thrombosisAntithrombin Antithrombin Manufactured in liverOther designationsHeparin cofactor I Antithrombin IIISerine protease inhibitorRequires heparan or heparin to functionHeparin requires antithrombin to functionAntithrombin Natural anticoagulant activityInactivates thrombin and factors Xa, IXa and XIaForms 1:1 complex with themDeficiency results in thrombosisVenous – more commonArterial Antithrombin DeficiencyEstimated frequency = 1%Types of deficiencyType I - quantitativeA = normal molecule, decreased amountB = abnormal molecule, abnormal amountType II – qualitative, abnormal moleculeA = activity and heparin binding siteB = activity onlyC = binding site onlyAntithrombin DeficiencyLaboratory testingAntithrombin activity or functionalChromogenic, if availableTreatmentAnticoagulationAntithrombin III concentrate (Thrombate III)Activation of Protein CProtein CManufactured in liverVitamin K dependentZymogen (must be activated)Thrombin combines with thrombomodulinRequires Protein S as cofactorProtein C DeficiencyNatural anticoagulant activityInactivates factors Va and VIIIaInactivates PAI-IDeficiency results in thrombosisVenous – more commonArterialPurpura fulminansHomozygous childrenPurpura FulminansProtein C DeficiencyEstimated frequency = 5%Types of deficiencyType I – quantitativeDecreased activity & decreased amountType II – qualitativeDecreased activityNormal amountProtein C DeficiencyLaboratory testingProtein C Activity or functionalChromogenic, if availableTreatmentAnticoagulationWarfarin-induced skin necrosis may occurProtein C concentrate Warfarin-induced Skin NecrosisProtein SManufactured in liver, endothelial cells, megakaryocytes, testes, kidney & brainVitamin K dependentMost of Protein S circulates bound to C4b binding proteinAcute phase reactantIncreases with inflammationProtein SFree Protein S is active formNatural anticoagulant activityCofactor for Protein CCombined with Protein CInactivates factors Va and VIIIaAloneWeak activity to inactivate factors Va, VIIIa and XaProtein S DeficiencyDeficiency results in thrombosisVenous – common with inactivityArterial – common in cerebral vesselsPurpura fulminansEstimated frequency = 3%Protein S DeficiencyTypes of deficiencyType I – quantitativeDecreased total, free and functionalType II – qualitativeNormal total and freeDecreased functionalType III – associated with increased C4bNormal totalDecreased free and functionalProtein S DeficiencyLaboratory testingProtein S activity or functionalProtein S freeC4b binding proteinChromogenic, if availableTreatmentAnticoagulationWarfarin-induced skin necrosisFactor XII Factor XIIManufactured in liverHageman factorSerine proteaseActivated by kallikrein (Fletcher Factor)Factor XIIa linked to kinin system, intrinsic fibrinolysis & complement systemsConverts plasminogen to plasminEstimated frequency of deficiency is lowFactor XII DeficiencyDeficiency decreases fibrinolysisDeficiency results in thrombosisVenous – more commonArterial – rareLaboratory testingFactor XII levelsTreatmentAnticoagulation Protein ZProtein ZVitamin K dependentCofactor for PZ-dependent protease inhibitor (PZI)Results in inactivation of factor XaFrequency of deficiency unknownDeficiency results in venous thrombosisLaboratory testing = Protein Z levelsTreatment = anticoagulationHeparin Cofactor IIHeparin Cofactor IIGlycoprotein, serine protease inhibitor familyActivated by heparan, heparin or dermatan sulfateResults in inhibition of thrombinNO effect on factor XaHeparin Cofactor II DeficiencyDecreased inhibition of thrombinDeficiency results in venous thrombosisActual risk is unknown at this timeLaboratory testingHeparin cofactor II levelsTreatmentAnticoagulation Tissue Factor Pathway Inhibitor (TFPI)TFPILocated in endothelial cellsBound to heparan sulfateHeparin increases plasma concentrationCombines with free factor Xa à Xa:TFPIXa:TFPI combines with VIIa:TFDecreased production of factor XInactivation of VIIa:TFTFPI DeficiencyFrequency unknownDeficiency causes:Venous thrombosisIntrauterine lethality (fetal loss)Neointimal proliferationLaboratory testingTFPI levelsTreatmentAnticoagulation Activated Protein C Resistance(APCR)APC ResistanceFrequency difficult to determine – may be as high as 30-50%Normal Protein C and Protein SNormal levels of activated Protein C with normal activityAPC unable to combine with factor Va for inactivationAPC ResistanceDefective factor VaReceptor for APC defectiveAPC unable to bind & inactivateNow looking at areas on factor VIIIaDefects in APC binding siteResults in venous & arterial thrombosisLaboratory testing = APC resistance assayTreatment = anticoagulation Inactivation of Factor VaFactor V LeidenMajor cause of APC ResistanceFrequency estimated at 20%Single point mutation (1691 Gà A) results in 506 Arg/Gln substitutionAffects first of three APC cleavage sitesAPC cannot down-regulate Factor VaFactor Va forms prothrombinase complex with factor Xa à conversion of prothrombin to thrombinProlonged procoagulant activityFactor V LeidenMost common genetic risk factor for thrombosisVenous and arterial Heterozygotes: 3 – 10X increased riskHomozygotes: 80X increased riskLaboratory testingRT-PCR for Factor V LeidenTreatment = anticoagulationOther Factor V MutationsFactor V Hong KongSingle point mutation results in 306 Arg/GlyRare cause of venous thrombosisFactor V CambridgeSingle point mutation results in 306 Arg/ThrRare cause of thrombosisFactor V LiverpoolSingle point mutation at Arg 679Rare cause of venous thrombosisHR2 HaplotypeSeveral mutationsRisk of venous thrombosis unknown at this timeThrombin: Central Role in HemostasisProthrombin G20210A MutationProthrombin G20210AEstimated frequency is 6%Mutation in location of 3’-UT regionResults in increased levels of ProthrombinVenous and possible arterial thrombosisHeterozygotes 3X increased risk of venous thrombosis Prothrombin G20210ALaboratory testingRT-PCR testing for gene mutationDo NOT measure factor II levelsTreatmentAnticoagulation Metabolism of HomocysteineHomocysteineTwo enzymesCystathionine B-synthaseConverts homocysteine to cystathionineDefect results in homocysteinuriaMTHFR (methylene tetrahydrofolate reductase)Converts homocysteine to methionineDefect causes homocysteinemia in presence of folate depletionThree vitamins – folate, B6 & B12HyperhomocysteinemiaElevated homocysteineDamage to endothelial cellsIncreased smooth muscle cell proliferationIncreased binding of Lp(a)Decreased vascular reactivity (decreased nitric oxide synthesis) Premature atherosclerosisIncreased tissue factor & factor Va releasedDecreased thrombomodulin (protein C activation) & heparan activityBlocked endothelial cell mediated fibrinolysis Venous/arterial thrombosisPregnancy-related vascular disorders = fetal lossHyperhomocysteinemiaLaboratory testingHomocysteine levelsMTHFR mutation by RT-PCRTreatmentVitamin supplementationFolate, B6 & B12Anticoagulation, if necessaryElevated Factor VIIIEstimated frequency is 16%Manufactured in endothelial cells & megakaryocytesAcute phase reactant with levels to ~400%Factors VIIIa & IXa form tenase complex (converts factor X to Xa)Factor Xa converts factor II to thrombinElevated Factor VIIILevels >400% are dangerousResult in increased thrombinIncrease in all factor VIII componentsVenous thrombosis occursPossible arterialLaboratory testing = Factor VIII levelsTreatment = anticoagulationOther Elevated Factor LevelsRisk factor for venous thrombosisLevels above the 90th percentileInclude factors XI, V and IIProbable genetic associationTestingSpecific factor levelTreatment = anticoagulationIncreased PAI-IPlasminogen Activator Inhibitor-IInhibits fibrinolysis (conversion of plasminogen to plasmin)Decreased fibrinolysis à increased and persistent clotting (venous thrombosis)Rare but may be cause when other tests are negativeTreatment = anticoagulationLABORATORY TESTINGWhen To TestHistory of recurrent thrombosisVTE prior to 45 years of ageUnprovoked VTE – any ageThrombosis in unusual sitesThrombosis + positive family history of thrombosisVTE secondary to pregnancy, OCT, hormone replacement therapyRecurrent fetal lossEarly onset atherosclerosisLaboratory TestingPattern the testing to the clinical presentationBe sure patient is clotting (not bleeding) before ordering tests for ThrombophiliaTry to test when patient is not acutely ill and not on anticoagulantsSeveral weeks after thrombotic eventSeveral weeks after anticoagulants discontinuedRecommended Lab TestingPT & aPTTNatural anticoagulants Antithrombin activityProtein C functionalProtein S functional & freeAPC resistanceFactor V LeidenProthrombin G20210AFactor VIIIFibrinogen Additional TestingHomocysteine levelMTHFROther factor levels (for increase)FibrinolysisPlasminogenPAI-IRecurrent Fetal LossAntiphospholipid SyndromeProtein C, S & ATIIIFactor V LeidenProthrombin G20210ACombinationPoints to RememberAvoid testing during acute eventUse functional assays firstConsider age & genderAlways confirm with repeat testingIf one abnormality identified, continue testing for others (combined common)Use PCR when available
Mar 22, 2007
Basic muscle pathalogy
Basic muscle pathalogy
Skeletal muscle:
Multiple nuclei are beneath the cell membrane (Sarcolemma).
It is normal in 3-5 % to see the nucleus in the center and more than that is pathologic.
Most of the cytoplasm is filled with Myofilaments .
Myofilament is constitute of myofibril which itself include the thin actin and thick myosin filaments.
Dystrophin is a sub sarcolemmal protein which can bind to Actin.
Sarcoplasmic reticulum and Z bands and T tubules are parallel to each other.
Multiple nuclei are beneath the cell membrane (Sarcolemma).
It is normal in 3-5 % to see the nucleus in the center and more than that is pathologic.
Most of the cytoplasm is filled with Myofilaments .
Myofilament is constitute of myofibril which itself include the thin actin and thick myosin filaments.
Dystrophin is a sub sarcolemmal protein which can bind to Actin.
Sarcoplasmic reticulum and Z bands and T tubules are parallel to each other.

Smooth mus:
Notice the elongated, "cigar-shaped" nuclei of the smooth muscle cells, the intercellular spaces between the smooth muscle cells, Don't expect every fiber in a section to exhibit a nucleus

Skeletal muscle stains:
First stain is the H&E (as the first picture above)
second is :

ATPase alkaline
type 2 dark
Checker board pattern
Fiber typing in denervation
type 2 dark
Checker board pattern
Fiber typing in denervation

ATPase acidic type 2 light
Checker board pattern
Fiber typing in denervation
Checker board pattern
Fiber typing in denervation

Trichrome:
mitochondrial ‘Red ragged fiber”
Rime vacuoles (in I.B.M.)
Nemaline rods (nemaline myopathy)
Subsarcolemma aggregation in channelopathies
mitochondrial ‘Red ragged fiber”
Rime vacuoles (in I.B.M.)
Nemaline rods (nemaline myopathy)
Subsarcolemma aggregation in channelopathies

NADH:
congenital Structural abnormality,target
fiber(central core
disease) or vacules and ..
congenital Structural abnormality,target
fiber(central core
disease) or vacules and ..

Acid phosphatase:
Mnemonic (acid chews up so acid for degeneration )
and it will look red. in opposite Alk phosphate shows regenerating fibers.
Also a good stain to trace the inflammation
and it will look red. in opposite Alk phosphate shows regenerating fibers.
Also a good stain to trace the inflammation

Alk phosphatase, Regenerating fiber will look blue
,nicely highlight the vessels too.
,nicely highlight the vessels too.

PAS: glycogen storage disease , type 1 darker (more glycogen)


SDH: Succinate dehydrogenase

TYPES OF MUSCLE FIBERS
Humans have basically three different types of muscle fibers.
A:Slow- twitch (ST or Type I) fibers are identified by a slow contraction time and a high resistance to fatigue. Structurally, they have a small motor neuron and fiber diameter, a high mitochondrial and capillary density, and
high myoglobin content, Energetically, they have a
low supply of creatine phosphate (a high-energy substrate used for quick, explosive movements), a low glycogen content, and
a wealthy store of triglycerides (the stored form of fat). Functionally, ST fibers are used for aerobic activities requiring low-level force production, such as walking and maintaining posture. Most activities of daily living use ST fibers.
B:Fast-twitch (FT or Type II) fibers:
The differences in the speeds of contraction that gives the fibers their names can be explained, in part, by the rates of release of calcium by the sarcoplasmic reticulum (the muscle's storage site for calcium) and by the activity of the enzyme (myosin-ATPase) that breaks down ATP inside the myosin head of the contractile proteins.
Both of these characteristics are faster and greater in the FT fibers (Fitts & Widrick, 1996; Harigaya & Schwartz, 1969).
Fast-twitch fibers are further divided into fast-twitch A (FT -A or Type IIA) and fast- twitch B (FT -B or Type lIB) fibers. FT -A fibers have a moderate resistance to fatigue and represent a transition between the two extremes of the ST and FT -B fibers. Structurally, FT -A fibers have a large motor neuron and fiber diameter, a high mitochondrial density, a medium capillary density, and a medium myoglobin content. They are high in creatine phosphate and glycogen and medium in triglyceride stores. They have both a high glycolytic and oxidative enzyme activity. Functionally, they are used for prolonged anaerobic activities with a relatively high force output, such as racing 400 meters.
Fast-twitch B fibers, on the other hand, are very sensitive to fatigue and are used for short anaerobic, high force production activities, such as sprinting, hurdling, jumping, and putting the shot. These fibers are also capable of producing more power than ST fibers. Like the FT -A fibers, FT -B fibers have a large motor neuron and fiber diameter, but a low mitochondrial and capillary density and myoglobin content. They also are high in creatine phosphate and glycogen, but low in triglycerides. They contain many glycolytic enzymes but few oxidative enzymes. Table 1 summarizes some major characteristics of the three fiber types.
Fast-twitch fibers are further divided into fast-twitch A (FT -A or Type IIA) and fast- twitch B (FT -B or Type lIB) fibers. FT -A fibers have a moderate resistance to fatigue and represent a transition between the two extremes of the ST and FT -B fibers. Structurally, FT -A fibers have a large motor neuron and fiber diameter, a high mitochondrial density, a medium capillary density, and a medium myoglobin content. They are high in creatine phosphate and glycogen and medium in triglyceride stores. They have both a high glycolytic and oxidative enzyme activity. Functionally, they are used for prolonged anaerobic activities with a relatively high force output, such as racing 400 meters.
Fast-twitch B fibers, on the other hand, are very sensitive to fatigue and are used for short anaerobic, high force production activities, such as sprinting, hurdling, jumping, and putting the shot. These fibers are also capable of producing more power than ST fibers. Like the FT -A fibers, FT -B fibers have a large motor neuron and fiber diameter, but a low mitochondrial and capillary density and myoglobin content. They also are high in creatine phosphate and glycogen, but low in triglycerides. They contain many glycolytic enzymes but few oxidative enzymes. Table 1 summarizes some major characteristics of the three fiber types.
RECRUITMENT OF MUSCLE FIBERS
Muscles produce force by recruiting motor units (a group of muscle fibers innervated by a motor neuron) along a gradient. During voluntary isometric and concentric contractions, the orderly pattern of recruitment is controlled by the size of the motor unit, a condition known as the size principle (Henneman, et al., 1974). Small motor units, which contain slow-twitch muscle fibers, have the lowest firing threshold and are recruited first. Demands for larger forces are met by the recruitment of increasingly larger motor units. The largest motor units that contain the fast-twitch B fibers have the highest threshold and are recruited last.
Mar 21, 2007
Arteriovenous malformations
Arteriovenous malformations
Vascular malformations
1-AVM(Arteriovenous malformations)
2-Cavernous hemangiomas
3-Venous angiomas
4-Capillary telangiectasias

Definition:represent an aberrant connection between the arterial and venous circulation with bypass of the capillary system .

AVM
This network of abnormal connections represents the "nidus"
AVM
Feeding arteries in the arteriovenous malformation consist of a high blood flow due to low resistance within the arteriovenous malformation.(no capillaries)
low resistance is thought to result in ischemic events, or the Steal phenomenon.
Steal phenomenon
Shunting of blood away from a nervous system location causing transient or persistent ischemia or infarction.
Yamada S, Cojocaru T. Arteriovenous malformations. In: Wood JH, editor. Cerebral blood flow: physiologic and clinical aspects. New York: McGraw-Hill, 1987:580-90.
Hoffman WE. Brain tissue oxygenation in patients with cerebral occlusive disease and arteriovenous malformations. Br J Anaesth 1997 Feb;78(2):169-71.
AVM
Etiology:
Most often these malformations are due to congenital anomalies
Other etiologic include
trauma
radiation.
AVM
Time course-Location:
Arteriovenous malformations evolve slowly over many years and can occur in any location
most often in MCA distribution and in the parietal or frontal lobes.
AVM
Female-male
Commonly present in 2nd and 3rd decade
tangle of dilated veins, filled with arterialized blood ("red veins") is evident, as is hyperemia of the cortical surface
AVM
Hemosiderin deposits :
Adjacent to the AVM can frequently be observed, even in patients who have no history of hemorrhage, indicating that blood leakage is common.
fibromuscular dysplasia and thrombosis
Clinical manifestations
most common presentation for AVM ?
?
3 cm
Hemorrhage typically occurs in arteriovenous malformations <>3 cm
AVM AND HEMORRHAGE
AVM and hemorrhage
Risk of hemorrhage in patients diagnosed with arteriovenous malformations who have not had a previous hemorrhage is in the range of 1.3% to 3.9% yearly
53% of patients with arteriovenous malformations will experience a hemorrhage
Hofmeister C, Stapf C, Hartmann, Stroke 2000 Jun;31(6):1307-10. Demographic, morphological, and clinical characteristics of 1289 patients with brain arteriovenous malformation.
AVM and hemorrhage
SAH
intraparenchymal
Small leaks (hemosiderin deposits)
Factors that Contribute to Increased Risk of Hemorrhage from an Arteriovenous Malformation
Clinical factors
History of hypertension
History of previous hemorrhage
Factors that Contribute to Increased Risk of Hemorrhage from an Arteriovenous Malformation
AVM and hemorrhage
Comparison of Risk Factors and Annual Risk of Hemorrhage Stroke 1996 Jan;27(1):1-6
AVM AND SEIZURE
3 cm
Seizures typically accompany arteriovenous malformations >3 cm
hemorrhage typically occurs in arteriovenous malformations <>6cm 3p
Eloquence of location,1p noneloquence 0
Deep Venous drainage present 1
Grades 1 to 3 are typically treated with microsurgery
Unruptured AVM
Higher grades AVM are more challenging and require multidisciplinary approach for treatment .
Embolization (not used grade1-3)
Stereotactic radiosurgery
SEIZURE
Treatment of lesions presenting with seizures includes surgical resection, embolization, radiosurgery, and anticonvulsant therapy.
SEIZURE
3 year minimum follow-up who had AVM and preoperative seizures, revealed
83% of patients who underwent AVM resection were seizure free (with 48% no longer receiving anticonvulsant therapy),
17% still suffered intermittent seizures
Seizure outcome in patients with surgically treated cerebral arteriovenous malformations. Piepgras , J Neurosurg 1993 Jan;78(1):5-11
Bradly: Ogilvy et al 2001,succ
AVM and Pregnancy
?
AVM and Pregnancy
Bradely:
ICH by AVM is as common as ICH with aneurysems
Time dosen’t always correlate with peak cardiovascular changes
Labor is highest risk period
Descision about Tx should be base on Neurosurgical rather Obstetric criteia
AVM and Pregnancy
Bradley:
Risk of rebreeding is higher in pregnancy
Elective cesarean section may carry the lowest risk
Cavernous hemangiomas
seizure is the most common presentation (in contrast in AVM hemorrhage)38% to 100% of patients
Focal neurologic deficits are the second most common clinical manifestation and have been reported to present in 15.4% to 46.6% of patients.
The incidence of hemorrhage has been estimated from a low of 0.1% to 1.1%
Venous angiomas
Seizures are the most common type of presentation in venous angiomas.
The overall rate of bleeding in venous angiomas has been reported to be between 1% and 16%
Infratentorial and deeply draining supratentorial have a higher incidence of hemorrhage and rebleeding than do superficial supratentorial venous angiomas
Venous angiomas most often occur in the subcortical areas of the frontal lobe or cerebellum
Vascular malformations
1-AVM(Arteriovenous malformations)
2-Cavernous hemangiomas
3-Venous angiomas
4-Capillary telangiectasias

Arteriovenous malformations
Definition:represent an aberrant connection between the arterial and venous circulation with bypass of the capillary system .

AVM
This network of abnormal connections represents the "nidus"
AVM
Feeding arteries in the arteriovenous malformation consist of a high blood flow due to low resistance within the arteriovenous malformation.(no capillaries)
low resistance is thought to result in ischemic events, or the Steal phenomenon.
Steal phenomenon
Shunting of blood away from a nervous system location causing transient or persistent ischemia or infarction.
Yamada S, Cojocaru T. Arteriovenous malformations. In: Wood JH, editor. Cerebral blood flow: physiologic and clinical aspects. New York: McGraw-Hill, 1987:580-90.
Hoffman WE. Brain tissue oxygenation in patients with cerebral occlusive disease and arteriovenous malformations. Br J Anaesth 1997 Feb;78(2):169-71.
AVM
Etiology:
Most often these malformations are due to congenital anomalies
Other etiologic include
trauma
radiation.
AVM
Time course-Location:
Arteriovenous malformations evolve slowly over many years and can occur in any location
most often in MCA distribution and in the parietal or frontal lobes.
AVM
Female-male
Commonly present in 2nd and 3rd decade
tangle of dilated veins, filled with arterialized blood ("red veins") is evident, as is hyperemia of the cortical surface
AVM
Hemosiderin deposits :
Adjacent to the AVM can frequently be observed, even in patients who have no history of hemorrhage, indicating that blood leakage is common.
fibromuscular dysplasia and thrombosis
Clinical manifestations
most common presentation for AVM ?
?
3 cm
Hemorrhage typically occurs in arteriovenous malformations <>3 cm
AVM AND HEMORRHAGE
AVM and hemorrhage
Risk of hemorrhage in patients diagnosed with arteriovenous malformations who have not had a previous hemorrhage is in the range of 1.3% to 3.9% yearly
53% of patients with arteriovenous malformations will experience a hemorrhage
Hofmeister C, Stapf C, Hartmann, Stroke 2000 Jun;31(6):1307-10. Demographic, morphological, and clinical characteristics of 1289 patients with brain arteriovenous malformation.
AVM and hemorrhage
SAH
intraparenchymal
Small leaks (hemosiderin deposits)
Factors that Contribute to Increased Risk of Hemorrhage from an Arteriovenous Malformation
Clinical factors
History of hypertension
History of previous hemorrhage
Factors that Contribute to Increased Risk of Hemorrhage from an Arteriovenous Malformation
AVM and hemorrhage
Comparison of Risk Factors and Annual Risk of Hemorrhage Stroke 1996 Jan;27(1):1-6
AVM AND SEIZURE
3 cm
Seizures typically accompany arteriovenous malformations >3 cm
hemorrhage typically occurs in arteriovenous malformations <>6cm 3p
Eloquence of location,1p noneloquence 0
Deep Venous drainage present 1
Grades 1 to 3 are typically treated with microsurgery
Unruptured AVM
Higher grades AVM are more challenging and require multidisciplinary approach for treatment .
Embolization (not used grade1-3)
Stereotactic radiosurgery
SEIZURE
Treatment of lesions presenting with seizures includes surgical resection, embolization, radiosurgery, and anticonvulsant therapy.
SEIZURE
3 year minimum follow-up who had AVM and preoperative seizures, revealed
83% of patients who underwent AVM resection were seizure free (with 48% no longer receiving anticonvulsant therapy),
17% still suffered intermittent seizures
Seizure outcome in patients with surgically treated cerebral arteriovenous malformations. Piepgras , J Neurosurg 1993 Jan;78(1):5-11
Bradly: Ogilvy et al 2001,succ
AVM and Pregnancy
?
AVM and Pregnancy
Bradely:
ICH by AVM is as common as ICH with aneurysems
Time dosen’t always correlate with peak cardiovascular changes
Labor is highest risk period
Descision about Tx should be base on Neurosurgical rather Obstetric criteia
AVM and Pregnancy
Bradley:
Risk of rebreeding is higher in pregnancy
Elective cesarean section may carry the lowest risk
Cavernous hemangiomas
seizure is the most common presentation (in contrast in AVM hemorrhage)38% to 100% of patients
Focal neurologic deficits are the second most common clinical manifestation and have been reported to present in 15.4% to 46.6% of patients.
The incidence of hemorrhage has been estimated from a low of 0.1% to 1.1%
Venous angiomas
Seizures are the most common type of presentation in venous angiomas.
The overall rate of bleeding in venous angiomas has been reported to be between 1% and 16%
Infratentorial and deeply draining supratentorial have a higher incidence of hemorrhage and rebleeding than do superficial supratentorial venous angiomas
Venous angiomas most often occur in the subcortical areas of the frontal lobe or cerebellum
Syndrome of continues motor unit activity
Syndrome of continues motor unit activity
Neuromyotonia
Neuromyotonia and myokymia
• The terms “neuromyotonia” and “myokymia” have both been used to describe “clinical phenomena” as well as distinct patterns of abnormal “electrical discharge” recorded during needle electromyography.
Neuromyotonia and myokymia
•This dual nomenclature has created confusion over the years, but no other set of clearer definitions has yet been universally accepted.
Neuromyotonia and myokymia
Both are related clinical phenomena that
result from hyperexcitability of peripheral
nerve motor axons.
Neuromyotonia and myokymia
•Whether they are really separate and distinct clinical entities or just reflect a difference in the severity of the same underlying electrophysiological abnormality remains undetermined.
Neuromyotonia and myokymia
Both may occur in a generalized or focal
fashion and reflect a generalized or
focal alteration in the microenvironment
or membrane of the peripheral nerve.
•Both clinical neuromyotonia and clinical myokymia are also classified as syndromes of continuous motor unit activity.
EMG Characteristics of Neuromyotonia
EMG Characteristics of Neuromyotonia
•Single MUAP firing rapidly
•150 Hz to 300 Hz discharges in long trains
•Trains occur at random intervals
•Train duration up to several seconds
•Decrementing train , Trains start and stop abruptly
•
EMG Characteristics of Myokymia
•Single MUAP firing as bursts of multiplets
•30 Hz to 40 Hz discharges in short bursts
•Burst occur at 2 Hz to 10 Hz
•Burst duration is 100 ms to 900 ms
•Semi-rhythmic burst pattern
•Bursts start and stop abruptly
Neuromyotonia
•is a syndrome marked by prominent and continuous muscle twitching and stiffness
• typically resulting from neuromyotonic and myokymic discharges.
Neuromyotonia
appear in adolescence and adult years. Diagnosis rests on both the clinical manifestations and typical electrophysiologic findings. Features of clinical neuromyotonia include:
Neuromyotonia
•clinical pseudomyotonia (slow muscle relaxation after a forceful contraction)
•contractures of the hands and feet (carpopedal spasms)
•Muscles of the limbs and trunk are stiff and rigid.
Clinical neuromyotonia
•Stiffness is more pronounced in the distal than proximal muscles, and it is worsened by exercise, although it may improve transiently with repetitive movement. Posture may be abnormal with exaggerated kyphosis, and movement is stiff and slow.
•Weight loss is common. The muscles may be well-developed, and sweating may be prominent, possibly because heat is generated by the excessive and constant muscle activity
ptosis, baldness, and temporalis muscle atrophy, cataractsTrinucleotide repeats , CTG .delayed muscle relaxation called myotoniapercussion."dive-bomber" activity that is the EMG hallmark of the disease.
•Dyspnea may result from tightening of the respiratory muscles. Bulbar and laryngeal muscles may be affected. The tongue and jaw become stiff, making swallowing difficult, and the voice turns hoarse
This abnormal activity persists during sleep .
•Focal neuromyotonic syndrome
•Morvan syndrome :
Acompanied with: confusion, insomnia, and hallucinations .
•Ocular neuromyotonia:
Includes intermittent diplopia produced by spasms of the extraocular muscles that occur spontaneously or in response to sustained eccentric gaze.
Physical examination
•Carpopedal spasms with flexion of the wrist, extension of the fingers, and plantar flexion of the feet .
•In clinical neuromyotonia demonstrates normal or depressed tendon reflexes, sometimes with coexisting sensorimotor peripheral neuropathy.
Laboratory studies
May demonstrate serum antibodies to voltage-gated potassium channels as well as mildly increased serum potassium . Oligoclonal bands have been reported in the cerebrospinal fluid .
Etiology
•Clinical neuromyotonia, in most cases, is autoimmune and sporadic .
•Many cases remain idiopathic
•A minority of neuromyotonic syndromes are hereditary and occur with some of the inherited neuropathies ( like the axonal variant of Charcot-Marie-Tooth disease).
•Antivoltage gated potassium channel antibodies are found in 40% to 50% of patients with acquired clinical neuromyotonia .(Vincent 2000; Gutmann 2001a; Van Parijs et al 2002)
•Clear association with thymoma, myasthenia gravis, lung cancer, and neuronal ganglionic antiacetylcholine receptor antibodies. (Vernino et al 1998; Hart et al 2002)
•Clinical neuromyotonia may also appear as a consequence of intoxication with mercury, penicillamine and oxaliplatin.
•Paraneoplastic syndromes of clinical neuromyotonia with hypernephroma and thymoma have also been reported, along with peripheral neuropathy, in bronchogenic carcinoma .
Treatment
•The clinical neuromyotonic syndromes may respond to immunomodulatory treatment including plasmapheresis, intravenous immunoglobulin, and steroids.
Treatment
•Neuromyotonia can sometimes be suppressed with phenytoin, carbamazepine, and dantrolene.
•In some patients, treatment can eventually be discontinued without recurrence , whereas others require life-long therapy.
•Clinical myokymia, in contrast, most commonly occurs as a component of other serious disorders .
•Focal myokymia may be caused by multiple sclerosis, pontine tumors, Guillain-Barré syndrome, radiation plexitis, and, rarely, rattlesnake envenomation. Facial myokymia may also be seen in Bell palsy, syringobulbia, polyradiculoneuropathy, central pontine myelinolysis, tuberculoma, meningeal carcinomatosi, meningeal sarcoidosis, lymphocytic meningoradiculitis, basilar invagination, phosgene poisoning, and hemifacial spasm. Facial myokymia can also occur spontaneously after brain death.
• Limb myokymia occurs in chronic inflammatory demyelinating polyneuropathy, rare compressive neuropathies, syringomyelia, spinal stenosis, conus medullaris teratoma, radiculopathy, neurocysticercosis, subarachnoid hemorrhage, and following cardiopulmonary arrest.
•Generalized myokymia may appear as a part of clinical neuromyotonia, restless leg syndrome, cramp fasciculation syndrome, gluten-sensitive enteropathy, and with clozapine use.
Neuromyotonia
Neuromyotonia and myokymia
• The terms “neuromyotonia” and “myokymia” have both been used to describe “clinical phenomena” as well as distinct patterns of abnormal “electrical discharge” recorded during needle electromyography.
Neuromyotonia and myokymia
•This dual nomenclature has created confusion over the years, but no other set of clearer definitions has yet been universally accepted.
Neuromyotonia and myokymia
Both are related clinical phenomena that
result from hyperexcitability of peripheral
nerve motor axons.
Neuromyotonia and myokymia
•Whether they are really separate and distinct clinical entities or just reflect a difference in the severity of the same underlying electrophysiological abnormality remains undetermined.
Neuromyotonia and myokymia
Both may occur in a generalized or focal
fashion and reflect a generalized or
focal alteration in the microenvironment
or membrane of the peripheral nerve.
•Both clinical neuromyotonia and clinical myokymia are also classified as syndromes of continuous motor unit activity.
EMG Characteristics of Neuromyotonia
EMG Characteristics of Neuromyotonia
•Single MUAP firing rapidly
•150 Hz to 300 Hz discharges in long trains
•Trains occur at random intervals
•Train duration up to several seconds
•Decrementing train , Trains start and stop abruptly
•
EMG Characteristics of Myokymia
•Single MUAP firing as bursts of multiplets
•30 Hz to 40 Hz discharges in short bursts
•Burst occur at 2 Hz to 10 Hz
•Burst duration is 100 ms to 900 ms
•Semi-rhythmic burst pattern
•Bursts start and stop abruptly
Neuromyotonia
•is a syndrome marked by prominent and continuous muscle twitching and stiffness
• typically resulting from neuromyotonic and myokymic discharges.
Neuromyotonia
appear in adolescence and adult years. Diagnosis rests on both the clinical manifestations and typical electrophysiologic findings. Features of clinical neuromyotonia include:
Neuromyotonia
•clinical pseudomyotonia (slow muscle relaxation after a forceful contraction)
•contractures of the hands and feet (carpopedal spasms)
•Muscles of the limbs and trunk are stiff and rigid.
Clinical neuromyotonia
•Stiffness is more pronounced in the distal than proximal muscles, and it is worsened by exercise, although it may improve transiently with repetitive movement. Posture may be abnormal with exaggerated kyphosis, and movement is stiff and slow.
•Weight loss is common. The muscles may be well-developed, and sweating may be prominent, possibly because heat is generated by the excessive and constant muscle activity
ptosis, baldness, and temporalis muscle atrophy, cataractsTrinucleotide repeats , CTG .delayed muscle relaxation called myotoniapercussion."dive-bomber" activity that is the EMG hallmark of the disease.
•Dyspnea may result from tightening of the respiratory muscles. Bulbar and laryngeal muscles may be affected. The tongue and jaw become stiff, making swallowing difficult, and the voice turns hoarse
This abnormal activity persists during sleep .
•Focal neuromyotonic syndrome
•Morvan syndrome :
Acompanied with: confusion, insomnia, and hallucinations .
•Ocular neuromyotonia:
Includes intermittent diplopia produced by spasms of the extraocular muscles that occur spontaneously or in response to sustained eccentric gaze.
Physical examination
•Carpopedal spasms with flexion of the wrist, extension of the fingers, and plantar flexion of the feet .
•In clinical neuromyotonia demonstrates normal or depressed tendon reflexes, sometimes with coexisting sensorimotor peripheral neuropathy.
Laboratory studies
May demonstrate serum antibodies to voltage-gated potassium channels as well as mildly increased serum potassium . Oligoclonal bands have been reported in the cerebrospinal fluid .
Etiology
•Clinical neuromyotonia, in most cases, is autoimmune and sporadic .
•Many cases remain idiopathic
•A minority of neuromyotonic syndromes are hereditary and occur with some of the inherited neuropathies ( like the axonal variant of Charcot-Marie-Tooth disease).
•Antivoltage gated potassium channel antibodies are found in 40% to 50% of patients with acquired clinical neuromyotonia .(Vincent 2000; Gutmann 2001a; Van Parijs et al 2002)
•Clear association with thymoma, myasthenia gravis, lung cancer, and neuronal ganglionic antiacetylcholine receptor antibodies. (Vernino et al 1998; Hart et al 2002)
•Clinical neuromyotonia may also appear as a consequence of intoxication with mercury, penicillamine and oxaliplatin.
•Paraneoplastic syndromes of clinical neuromyotonia with hypernephroma and thymoma have also been reported, along with peripheral neuropathy, in bronchogenic carcinoma .
Treatment
•The clinical neuromyotonic syndromes may respond to immunomodulatory treatment including plasmapheresis, intravenous immunoglobulin, and steroids.
Treatment
•Neuromyotonia can sometimes be suppressed with phenytoin, carbamazepine, and dantrolene.
•In some patients, treatment can eventually be discontinued without recurrence , whereas others require life-long therapy.
•Clinical myokymia, in contrast, most commonly occurs as a component of other serious disorders .
•Focal myokymia may be caused by multiple sclerosis, pontine tumors, Guillain-Barré syndrome, radiation plexitis, and, rarely, rattlesnake envenomation. Facial myokymia may also be seen in Bell palsy, syringobulbia, polyradiculoneuropathy, central pontine myelinolysis, tuberculoma, meningeal carcinomatosi, meningeal sarcoidosis, lymphocytic meningoradiculitis, basilar invagination, phosgene poisoning, and hemifacial spasm. Facial myokymia can also occur spontaneously after brain death.
• Limb myokymia occurs in chronic inflammatory demyelinating polyneuropathy, rare compressive neuropathies, syringomyelia, spinal stenosis, conus medullaris teratoma, radiculopathy, neurocysticercosis, subarachnoid hemorrhage, and following cardiopulmonary arrest.
•Generalized myokymia may appear as a part of clinical neuromyotonia, restless leg syndrome, cramp fasciculation syndrome, gluten-sensitive enteropathy, and with clozapine use.
Mar 5, 2007
Fibromuscular dysplasia
91 year old female was admitted for left sided weakness secondary to right MCA infarction. On CT angiogram she has the typical “string-of-beads” stenoses of the bilateral internal carotid artery.

Feb 24, 2007
WEBINO
Wall-eyed bilateral internuclear ophthalmoplegia

82 year old right handed male with history of Giant cell artritis presented in our ED with 2 days compliants of falls and double vision. On exam demonstrated bilateral internuclear ophthalmoplegia.

82 year old right handed male with history of Giant cell artritis presented in our ED with 2 days compliants of falls and double vision. On exam demonstrated bilateral internuclear ophthalmoplegia.

Jan 26, 2007
Olivopontocerebellar atrophy
Olivopontocerebellar atrophy (OPCA)

This is MRI of a 50 year old female with Family history of Olivopontocerebellar atrophy in father. She has been suffering from ataxias and falls for 3 years. On exam demonstrate dysmetric finger to nose and heel to shin, has hypo metric saccades , mild ocular apraxia . There is lack of Bulbar weakness or other parkinsonism features.

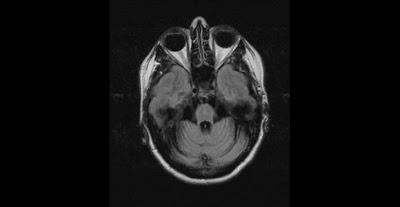

This is MRI of a 50 year old female with Family history of Olivopontocerebellar atrophy in father. She has been suffering from ataxias and falls for 3 years. On exam demonstrate dysmetric finger to nose and heel to shin, has hypo metric saccades , mild ocular apraxia . There is lack of Bulbar weakness or other parkinsonism features.



Jan 23, 2007
Bilateral grey matter lesions


Symmetric Lesions of the Deep Gray Nucl
1 Acute:
•Toxic
•Metabolic
•Hypoxic
•Vascular
•infectious
•unknown
2-Chronic
Develop over months to years.
Heredodegenerative,
Infectious
Indolent metabolic and toxic
Neoplastic
Unknown pathophysiology.
Symmetric Lesions of the Deep Gray Nuclei
Symmetric Lesions of the Deep Gray Nuclei
Neurologic presentations
1-Acute
Manifest as alterations in mental status that include behavioral change, somnolence,and coma.
involuntary movements and seizure
Neurologic presentations (acute)
Seizures, while less common, may be an initial sign.
Seizures occur as a result of associated cortical involvement from the underlying etiology affecting the DGMN.
Neurologic presentations
Manifest as alterations in mental status that include behavioral change, somnolence,and coma.
involuntary movements and seizure
Neurologic presentations (acute)
Seizures, while less common, may be an initial sign.
Seizures occur as a result of associated cortical involvement from the underlying etiology affecting the DGMN.
Neurologic presentations
2-chronic
Chronic conditions produce a more delayed evolution of additional clinical
signs, such as involuntary movements, altered tone (rigidity/cogwheeling), and other Parkinsonian features
History
hypertension
Question about toxic, metabolic, and hypoxic insults
suicide attempt
exposure to neurotoxins that include cyanide, methanol, and carbon monoxide
substance abuse
Chronic conditions produce a more delayed evolution of additional clinical
signs, such as involuntary movements, altered tone (rigidity/cogwheeling), and other Parkinsonian features
History
hypertension
Question about toxic, metabolic, and hypoxic insults
suicide attempt
exposure to neurotoxins that include cyanide, methanol, and carbon monoxide
substance abuse
History
Metabolic disease, can manifest as an acute intoxication with lethargy or seizures and less commonly with focal neurologic deficit.
In each circumstance multiorgan involvement is possible and often acute and catastrophic.
in chronic cases a history related to
nutrition; alcohol use; chronic toxic exposure
brain radiation therapy;
conditions associated with liver disease;
prior streptococcal infection;
HIV infection or risk factors;
family history of :
Stroke,movement disorder
Dementia,neurodegenerative disease
Laboratory Investigations
Acute:
complete blood count
electrolytes investigations
liver and renal function tests, blood
glucose, blood gas
mycoplasma titers,
urine and blood toxicological studies
Laboratory Investigations
complete blood count
electrolytes investigations
liver and renal function tests, blood
glucose, blood gas
mycoplasma titers,
urine and blood toxicological studies
Laboratory Investigations
Chronic:
Routine blood and urine studies
liver function tests, serum calcium, ceruloplasmin, and parathyroid hormone assay
Testing for HIV and antistreptolysin titers
Testing for methylmalonic acid, skin fibroblast analysis of glutaryl-CoA dehydrogenase, and 24-hour urine collection for copper .
Laboratory Investigations
If considering Prion disease, an EEG and CSF for protein 14-3-3 should be obtained.
Specific clinical examination and genetic tests for neurofibromatosis type 1, Huntington’s disease, Tay-Sachs disease,
and mitochondrial disorders should also be considered.
Neuroimaging
Case :
A 30-year-old man with history of
suacidal attempts was brought to a
local emergency room apneic and
cyanotic and awoke over the next
12 days. After several months he made
a near full recovery with mild residual
dysarthria and athetoid movements of the upper extremities.
A subsequent CT scan showed bilateral symmetric lesions of the globus pallidus
Cyanide poisoning :
most commonly results
from homicide or suicide attempts with death resulting within minutes in 95% of cases. Inhibition of the mitochondrial cytochrome oxidase system, which blocks utilization of oxygen during oxidative phosphorylation, results in cytotoxic hypoxia.
Carbon Monoxide Poisoning
A 28-year-old woman was found unresponsive
in a closed garage with the car running during a suicide
attempt. On arrival to the hospital she was treated
with hyperbaric oxygen, carboxyhemoglobin level
Routine blood and urine studies
liver function tests, serum calcium, ceruloplasmin, and parathyroid hormone assay
Testing for HIV and antistreptolysin titers
Testing for methylmalonic acid, skin fibroblast analysis of glutaryl-CoA dehydrogenase, and 24-hour urine collection for copper .
Laboratory Investigations
If considering Prion disease, an EEG and CSF for protein 14-3-3 should be obtained.
Specific clinical examination and genetic tests for neurofibromatosis type 1, Huntington’s disease, Tay-Sachs disease,
and mitochondrial disorders should also be considered.
Neuroimaging
Case :
A 30-year-old man with history of
suacidal attempts was brought to a
local emergency room apneic and
cyanotic and awoke over the next
12 days. After several months he made
a near full recovery with mild residual
dysarthria and athetoid movements of the upper extremities.
A subsequent CT scan showed bilateral symmetric lesions of the globus pallidus
Cyanide poisoning :
most commonly results
from homicide or suicide attempts with death resulting within minutes in 95% of cases. Inhibition of the mitochondrial cytochrome oxidase system, which blocks utilization of oxygen during oxidative phosphorylation, results in cytotoxic hypoxia.
Carbon Monoxide Poisoning
A 28-year-old woman was found unresponsive
in a closed garage with the car running during a suicide
attempt. On arrival to the hospital she was treated
with hyperbaric oxygen, carboxyhemoglobin level
Leigh disease
Abnormalities of respiration are typical of Leigh disease and consist of periodic hyperventilation, apnea, gasping, sighing, and irregular breathing. Respiratory failure in Leigh disease often causes death.
Evaluation should include measurement of serum lactate/pyruvate, CSF lactate, and MRI.
If the diagnosis is in doubt, skin fibroblast studies
Muscle biopsy characteristically does not show ragged-red fibers and is normal
Abnormalities of respiration are typical of Leigh disease and consist of periodic hyperventilation, apnea, gasping, sighing, and irregular breathing. Respiratory failure in Leigh disease often causes death.
Evaluation should include measurement of serum lactate/pyruvate, CSF lactate, and MRI.
If the diagnosis is in doubt, skin fibroblast studies
Muscle biopsy characteristically does not show ragged-red fibers and is normal
Kearns-Sayre syndrome
the predominant clinical features are found in the central nervous system, skeletal muscle, and heart
obligatory triad of (1) onset before age 20, (2) pigmentary retinopathy, and (3) progressive external ophthalmoplegia.
In addition, at least 1 of the following must be present: (1) cardiac conduction block, (2) cerebrospinal fluid protein greater than 100 mg/dL, or (3) cerebellar ataxia
Kearns-Sayre syndrome
Other clinical manifestations seen in the majority of Kearns-Sayre syndrome patients include short stature, sensorineural hearing loss, impaired intellect, and limb weakness.
Kearns-Sayre syndrome
Elevated lactate and pyruvate
ECG must be performed to screen for heart block.
Lumbar puncture typically shows elevated CSF protein (usually over 100 mg/dL) with or without abnormal numbers of white blood cells.
Oligoclonal bands in the cerebrospinal fluid may be a nonspecific abnormality.
Kearns-Sayre syndrome
Ragged-red fibers on modified Gomori trichrome stain are the hallmark histological feature.
DHE protocol
Decadron 4
Reglan 10
DHE 1
(TEST .5 THE FIVE MINUTE LATER THE REST with base line EKG )
continue 24 h after headache is treated.
Reglan 10
DHE 1
(TEST .5 THE FIVE MINUTE LATER THE REST with base line EKG )
continue 24 h after headache is treated.
Dec 18, 2006
Sub acute combined degeneration (B12 deficency)


B12 deficiency
Sub acute combined degeneration
Case :66 yo female vegetarian with 6 month history off distal extremities parasthesia accompany by 4 month complaint of ataxia and memory loss.
HGB : 10.4 g/dL (11.2-15.2)
MCV : 117 Fl (78-100)
MCH : 41.7 pg(27.4-33.0)
Homocysteine total : 59 (4 to 14)
Methylmalonic Acid (pending)
RBC morphology : macrocyosis,Polychromasia,Anisocytosis
B12 : 190 pg/mL (211-911)
Folate : 20.6 ng/mL
Macrocytic anemi
Polymorph
Hypersegmented
neutrophiles
HGB : 10.4 g/dL (11.2-15.2)
MCV : 117 Fl (78-100)
MCH : 41.7 pg(27.4-33.0)
Homocysteine total : 59 (4 to 14)
Methylmalonic Acid (pending)
RBC morphology : macrocyosis,Polychromasia,Anisocytosis
B12 : 190 pg/mL (211-911)
Folate : 20.6 ng/mL
Macrocytic anemi
Polymorph
Hypersegmented
neutrophiles

Clinical Manifestation :
Hematologic
Neurologic
Neuropathy is symmetrical and affects the legs more than the arms.
It begins with paresthesias and ataxia associated with loss of vibration and position sense, and can progress to severe weakness, spasticity, clonus, paraplegia
Psychiatric disturbances, and dementia
Ischemic stroke
Optic neuropathy

Spongy or "vacuolar" degeneration of the spinal cord white matter
Management
4 injections of cyanocobalamin 1000 µg intramuscularly over 2 weeks.
In patients with pernicious anemia, monthly intramuscular injections of 1000 µg of cyanocobalamin
Dec 9, 2006
Guillain-Barré syndrome
Guillain-Barré syndrome
G.B.S
Acute inflammatory demyelinating polyradiculoneuropathy
A.I.D.P
French army neurologists in 1916
G. Guillain and J.A. Barré
Albuminocytologic dissociation
Etiology
Cell-mediated autoimmune disease of the peripheral nerves.
Nature of the antigens in acute Inflammatory demyelinating polyradiculoneuropathy is not known.
Variants of Guillain-Barré syndrome
CSF.Tx
1-Acute inflammatory demyelinating polyneuropathy
Symptoms and exam
Tendon reflexes
Severity
Evolution (1)
Autonomic function. Sixty-five percent of cases in some series have had manifestations of dysautonomia Sinus tachycardia occurs in more than 50% of severe cases (1).
EMG,CSF:
1-Ropper et al 1991
2-Acute motor-sensory axonal neuropathy
History: since 1916
Clinical picrure
Distinction from A.I.D.P
3-Acute motor axonal neuropathy (Chinese paralytic syndrome)
Clinical picture without sensory loss
Distinction from A.I.D.P
Campylobacter infection
4-Fisher syndrome
Clinical picture:
Characterized by 3 findings?
4-Fisher syndrome
Clinical picture: ophthalmoplegia, ataxia, areflexia
Also characterized by association with anti-GQ1b antibody
5-Sensory Guillain-Barré syndrome
6-pharyngeal-cervical-brachial variant
Limb muscles are spared ,manifests with acute oropharyngeal, neck, and shoulder weakness
7-Pandysautonomia
Acute or sub acute sympathetic and parasympathetic autonomic dysfunction with relative or complete preservation of somatic motor and sensory functions.
CSF, EMG, Autonomic study
Ganglionic acetylcholine receptors antibodies
Epidemiology
AIDP all parts of the world and at all ages
AIDP incidence is 0.6 to 2.4 cases per 100,000 population per year
Antecedent events
More than 50% AIDP follow by an infection
Other associated with AIDP include vaccination, surgery, epidural anesthesia, thrombolytic agents, and heroin use.
Diagnostic workup
CSF About 1 week after the onset of symptoms, the protein levels rise, reaching a peak in 3 to 4 weeks
Anti-GQ1b antibody
EMG
EMG
Electrophysiologic studies reveal a predominance of demyelinating features such as multifocal conduction block, slowing of nerve conduction velocities with prolonged distal and F-wave latencies, and temporal dispersion .
Temporal dispersion
EMG
Conduction studies are frequently normal at the beginning.
What might be the first change in EMG ?
Management
Corticosteroids randomized controlled trials revealed no benefit from such treatment (Hughes 1991)
Intravenous immunoglobulin, Plasmapheresis
Prevention of complications, such as respiratory failure Physical therapy
G.B.S
Acute inflammatory demyelinating polyradiculoneuropathy
A.I.D.P
French army neurologists in 1916
G. Guillain and J.A. Barré
Albuminocytologic dissociation
Etiology
Cell-mediated autoimmune disease of the peripheral nerves.
Nature of the antigens in acute Inflammatory demyelinating polyradiculoneuropathy is not known.
Variants of Guillain-Barré syndrome
CSF.Tx
1-Acute inflammatory demyelinating polyneuropathy
Symptoms and exam
Tendon reflexes
Severity
Evolution (1)
Autonomic function. Sixty-five percent of cases in some series have had manifestations of dysautonomia Sinus tachycardia occurs in more than 50% of severe cases (1).
EMG,CSF:
1-Ropper et al 1991
2-Acute motor-sensory axonal neuropathy
History: since 1916
Clinical picrure
Distinction from A.I.D.P
3-Acute motor axonal neuropathy (Chinese paralytic syndrome)
Clinical picture without sensory loss
Distinction from A.I.D.P
Campylobacter infection
4-Fisher syndrome
Clinical picture:
Characterized by 3 findings?
4-Fisher syndrome
Clinical picture: ophthalmoplegia, ataxia, areflexia
Also characterized by association with anti-GQ1b antibody
5-Sensory Guillain-Barré syndrome
6-pharyngeal-cervical-brachial variant
Limb muscles are spared ,manifests with acute oropharyngeal, neck, and shoulder weakness
7-Pandysautonomia
Acute or sub acute sympathetic and parasympathetic autonomic dysfunction with relative or complete preservation of somatic motor and sensory functions.
CSF, EMG, Autonomic study
Ganglionic acetylcholine receptors antibodies
Epidemiology
AIDP all parts of the world and at all ages
AIDP incidence is 0.6 to 2.4 cases per 100,000 population per year
Antecedent events
More than 50% AIDP follow by an infection
Other associated with AIDP include vaccination, surgery, epidural anesthesia, thrombolytic agents, and heroin use.
Diagnostic workup
CSF About 1 week after the onset of symptoms, the protein levels rise, reaching a peak in 3 to 4 weeks
Anti-GQ1b antibody
EMG
EMG
Electrophysiologic studies reveal a predominance of demyelinating features such as multifocal conduction block, slowing of nerve conduction velocities with prolonged distal and F-wave latencies, and temporal dispersion .
Temporal dispersion
EMG
Conduction studies are frequently normal at the beginning.
What might be the first change in EMG ?
Management
Corticosteroids randomized controlled trials revealed no benefit from such treatment (Hughes 1991)
Intravenous immunoglobulin, Plasmapheresis
Prevention of complications, such as respiratory failure Physical therapy
May 27, 2006
Hypoxic-ischemic encephalopathy
Anoxic-ischemic coma
Historical note
It was named "oxygen" (acid-former) by Antoine Lavoisier (1743-1794) of France
He made the greatest medical discoveries concerning oxygen's role in respiration.
In animal experiments, Lavoisier and others discovered that anoxia led to collapse and death.
In 1920 Barcroft introduced the terms "anoxic," "anemic," "histotoxic," and "stagnant" to designate the various forms of anoxia.
Barcroft J. Anoxemia. Lancet 1920;2:485-9.
Etiology
The brain utilizes oxygen to metabolize glucose. It cannot store oxygen and survives only for minutes after its oxygen supply is reduced below critical levels
Most vulnerable
1-Pyramidal cells of the third and fifth layers of the cerebral cortex
2-Pyramidal cells in Sommer sector of the hippocampus
3-Purkinje cells of the cerebellum
1-Pyramidal cells of the third and fifth layers of the cerebral cortex
3-Purkinje cells of the cerebellum all output of the cerebellum cortex carried by it to white matter
Lack of oxygen to the brain can be divided into :
Anoxic anoxia
Anemic anoxia
Ischemia anoxia
Ischemic anoxia
Describes a state of insufficient cerebral blood flow. "Low-flow states" may be secondary to cardiovascular collapse or conditions of increased vascular resistance as in stroke or migraine.
Anoxic anoxia
consists of low arterial oxygen content and tension. This may be secondary to decreased oxygen in the environment or inability for oxygen to enter the circulatory system as in pulmonary disease (P.E.).
Anemic anoxia
Consists of low oxygen content in the blood secondary to decreased hemoglobin content.
Brain normally consumes approximately 3.5 mL of oxygen for each 100 g of brain tissue per minute.
When this rate declines to 2.5 mL, delirium supervenes.
Rates of cerebral oxygen metabolism below 2 mL/100 g per minute are incompatible with a conscious state.
Epidemiology
Most of the time acute anoxic coma is a result of cardiac arrest .
Approximately 1.5 million people per year in the United States succumb to a cardiac death
Epidemiology
About 30% of the cardiac resuscitations attempted each year are considered "successful,"
Only 10% to 20% of survivors are able to resume their former lifestyles(Denton and Thomas 1997)
Epidemiology
Following cardiac arrest, individuals admitted to intensive care units in coma suffer a high mortality rate
Patients in coma for more than 48 hours have a 77% mortality rate By contrast, individuals not in coma have an 11% mortality rate (Teres et al 1982)
Prevention
Complications, such as hypoxic-ischemic encephalopathy, are closely linked to the time until the first defibrillatory shock is administered during cardiopulmonary resuscitation.
Increased survival rates with minimal complications are attained by reducing the time to initiation of cardiopulmonary resuscitation and defibrillation
In some cases, this can triple the chance of surviving a cardiac arrest outside of the hospital (Herlitz et al 1994)
Differential diagnosis
Severe degrees of anemia, congestive heart failure, hypotension, and pulmonary disease can lead to coma.
Occlusion of the pulmonary arteries during a pulmonary embolism produces an abrupt drop in cardiac output and cerebral blood flow.
Differential diagnosis
Fat embolism wherein pulmonary and cerebral vessels become occluded with lipid and fibrin debris can produce cerebral ischemia.
Anoxic coma also occurs in association with cardiac surgery (Hypotension).
Differential diagnosis
Hypertensive encephalopathy leads to diffuse cerebral ischemia. Alterations of the blood-brain barrier, arteriole necrosis, and diffuse infarcts and hemorrhages are present in this syndrome.
Differential diagnosis
Migraine headaches can result in coma. The mechanism is believed secondary to vascular insufficiency of the tributaries of the basilar artery.
Symptoms range from confusion, fever, meningismus, ataxia, syncope, and amnesia, to coma. Most episodes of coma resolve within 24 hours, but can persist for days and can be precipitated by head trauma, work, and angiography.
-Familial migraine coma: a case study Journal of Neurology February 1990 Issue: Volume 237, Number 1
-Coma associated with migraine .Rev Neurol. 1999 Dec 1-15;29(11):1048-51
Basilar migraine:
Usually during childhood and teenage years
Throbbing Occipital headache
Aura 10-40 minutes, visual changes
Tingling and numbness of hand and feet's
Ataxia of gait and speech, vertigo and tinnitus
Involvement of brainstem reticular formation can cause loss of consciousness
Bradley 2075
Diagnostic workup
…
CT or MRI are useful in determining the etiology of coma. These studies can differentiate between an ischemic infarct, intracerebral hemorrhage, and a mass lesion involving the cortex or the brainstem
Diagnostic workup
CT scan and global hypoxic injury :
There is a loss and reversal of normal grey-white matter differentiation with expanded grey matter showing lower attenuation. The basal ganglia are not sharply defined. There is also generalized effacement of cerebral sulci.
Diagnostic workup
MRI with the apparent diffusion coefficient of water is becoming a sensitive tool of neuronal physiology and may represent a reliable indicator of progressive neuronal injury following cerebral ischemia.
White matter demyelization and necrosis
Sub cortical u fibers
Corpus calusum
Int/ext capsules
Gray matter
Neuroradiology, Grossman 353-4
Diagnostic workup
EEG can be a sensitive indicator of cerebral function.
the EEG can be used to assess cerebral function and to guide therapy.
Coma Patterns
Alpha Coma, Burst Suppression
Periodic spiking , Triphasic waves
Electrocerebral inactivity
Diagnostic workup
EEG has been used to assess prognosis. In patients with terminal coma, onset of abnormal EEG changes may sometimes be suggestive of a poor outcome :
(Rothstein et al 1991)
Burst suppression pattern
Periodic pattern( Generalized periodic discharges, periodic lateralized epileptiform discharges.)
Diagnostic workup
BURST SUPRESSION
Diagnostic workup
Periodic lateralizing epileptiform discharges may occur following focal cerebral insults such as infarction, but do not necessarily indicate active epileptic activity.
Diagnostic workup
Alpha coma is frequently associated with cardiac arrest, but its prognostic significance is unclear.
EEG pattern does not change with stimulation.
( ? Extremely poor prognosis PEDLEY,358)
Diagnostic workup
If seizures develop and aggressive management is desired for the patient, then epileptic activity should be treated to reduce further neuronal cell loss in the cortex. Status epilepticus, status myoclonus, and myoclonic statusepilepticus are associated with inability to recover consciousness in cardiac arrest survivors (Krumholz et al 1988)
ICU? – Consult ! – Prognosis?
Prognosis and complications
Prediction of outcome following coma has been the subject of multiple clinical studies
Earlier reports have noted that post-arrest coma longer than 3 days carried an unfavorable prognosis (Bell and Hodgson 1974)
Prognosis and complications
Several studies even report some permanent neurologic sequelae if coma duration is greater than 6 hours (Maiese et al 1988)
Others have correlated outcome with the pattern of motor responses and the presence or absence of particular brainstem reflexes.
( Levy et al , predicting outcome from hypoxemic-ischemic coma 1985)
Levy examined 210 patients in whom cardiopulmonary failure was the cause of coma and correlated clinical signs with outcome at 1 year .
Patient age, sex, location of the initial insult, etiology of the coma, and the presence of generalized seizures did not influence the degree of recovery.
Levy et al 1985
In contrast, the clinical examination did correlate with recovery. Individuals without pupillary light reflexes at the initial examination never regained independence. Absence of corneal reflexes following the first day was also a poor prognosis.
Levy et al 1985
Prognosis and complications
Increased numbers of brainstem reflex abnormalities were associated with reduced survival
Snyder et al 1977 ,Rudolf et al 2000
The most favorable sign of a good outcome
(can be rare at day 1):
Any form of speech, orienting spontaneous eye movements, intact oculocephalic or oculovestibular responses, ability to follow commands, and normal skeletal tone.
Prognosis and complications
Additional investigations seek to identify cellular markers that may signal a poor neurologic prognosis, such as elevated levels of S-100B and interleukin-8 ,(Mussack et al 2002)
Mussack et al 2002
Serum S-100B and interleukin-8 as predictive markers for comparative neurologic outcome analysis of patients after cardiac arrest and severe traumatic brain injury
Significantly elevated S-100B and interleukin-8 serum levels 12 hrs after cardiac arrest suggest that primary brain damage and systemic inflammatory response are comparably serious with that of traumatic brain injury. In both collectives, increased S-100B values measured 12 hrs after insult correlated well with an unfavorable neurologic outcome after 12 months.
Decorticate or flexor responses occur after damage to the hemispheres, or in cases of diffuse depression of cortical function following cerebral ischemia. Decerebrate or extensor responses correlate with destructive lesions of the midbrain and upper pons, but also may be present in anoxic encephalopathy. The absence of motor response, especially if flaccidity and areflexia are present, indicates severe brainstem depression and is frequently found in terminal coma or in severe sedative intoxication. Withdrawal and localizing responses imply purposeful or voluntary behavior. Obeying commands is the best response and marks the return of consciousness.
Anoxic-ischemic coma
Historical note
It was named "oxygen" (acid-former) by Antoine Lavoisier (1743-1794) of France
He made the greatest medical discoveries concerning oxygen's role in respiration.
In animal experiments, Lavoisier and others discovered that anoxia led to collapse and death.
In 1920 Barcroft introduced the terms "anoxic," "anemic," "histotoxic," and "stagnant" to designate the various forms of anoxia.
Barcroft J. Anoxemia. Lancet 1920;2:485-9.
Etiology
The brain utilizes oxygen to metabolize glucose. It cannot store oxygen and survives only for minutes after its oxygen supply is reduced below critical levels
Most vulnerable
1-Pyramidal cells of the third and fifth layers of the cerebral cortex
2-Pyramidal cells in Sommer sector of the hippocampus
3-Purkinje cells of the cerebellum
1-Pyramidal cells of the third and fifth layers of the cerebral cortex
3-Purkinje cells of the cerebellum all output of the cerebellum cortex carried by it to white matter
Lack of oxygen to the brain can be divided into :
Anoxic anoxia
Anemic anoxia
Ischemia anoxia
Ischemic anoxia
Describes a state of insufficient cerebral blood flow. "Low-flow states" may be secondary to cardiovascular collapse or conditions of increased vascular resistance as in stroke or migraine.
Anoxic anoxia
consists of low arterial oxygen content and tension. This may be secondary to decreased oxygen in the environment or inability for oxygen to enter the circulatory system as in pulmonary disease (P.E.).
Anemic anoxia
Consists of low oxygen content in the blood secondary to decreased hemoglobin content.
Brain normally consumes approximately 3.5 mL of oxygen for each 100 g of brain tissue per minute.
When this rate declines to 2.5 mL, delirium supervenes.
Rates of cerebral oxygen metabolism below 2 mL/100 g per minute are incompatible with a conscious state.
Epidemiology
Most of the time acute anoxic coma is a result of cardiac arrest .
Approximately 1.5 million people per year in the United States succumb to a cardiac death
Epidemiology
About 30% of the cardiac resuscitations attempted each year are considered "successful,"
Only 10% to 20% of survivors are able to resume their former lifestyles(Denton and Thomas 1997)
Epidemiology
Following cardiac arrest, individuals admitted to intensive care units in coma suffer a high mortality rate
Patients in coma for more than 48 hours have a 77% mortality rate By contrast, individuals not in coma have an 11% mortality rate (Teres et al 1982)
Prevention
Complications, such as hypoxic-ischemic encephalopathy, are closely linked to the time until the first defibrillatory shock is administered during cardiopulmonary resuscitation.
Increased survival rates with minimal complications are attained by reducing the time to initiation of cardiopulmonary resuscitation and defibrillation
In some cases, this can triple the chance of surviving a cardiac arrest outside of the hospital (Herlitz et al 1994)
Differential diagnosis
Severe degrees of anemia, congestive heart failure, hypotension, and pulmonary disease can lead to coma.
Occlusion of the pulmonary arteries during a pulmonary embolism produces an abrupt drop in cardiac output and cerebral blood flow.
Differential diagnosis
Fat embolism wherein pulmonary and cerebral vessels become occluded with lipid and fibrin debris can produce cerebral ischemia.
Anoxic coma also occurs in association with cardiac surgery (Hypotension).
Differential diagnosis
Hypertensive encephalopathy leads to diffuse cerebral ischemia. Alterations of the blood-brain barrier, arteriole necrosis, and diffuse infarcts and hemorrhages are present in this syndrome.
Differential diagnosis
Migraine headaches can result in coma. The mechanism is believed secondary to vascular insufficiency of the tributaries of the basilar artery.
Symptoms range from confusion, fever, meningismus, ataxia, syncope, and amnesia, to coma. Most episodes of coma resolve within 24 hours, but can persist for days and can be precipitated by head trauma, work, and angiography.
-Familial migraine coma: a case study Journal of Neurology February 1990 Issue: Volume 237, Number 1
-Coma associated with migraine .Rev Neurol. 1999 Dec 1-15;29(11):1048-51
Basilar migraine:
Usually during childhood and teenage years
Throbbing Occipital headache
Aura 10-40 minutes, visual changes
Tingling and numbness of hand and feet's
Ataxia of gait and speech, vertigo and tinnitus
Involvement of brainstem reticular formation can cause loss of consciousness
Bradley 2075
Diagnostic workup
…
CT or MRI are useful in determining the etiology of coma. These studies can differentiate between an ischemic infarct, intracerebral hemorrhage, and a mass lesion involving the cortex or the brainstem
Diagnostic workup
CT scan and global hypoxic injury :
There is a loss and reversal of normal grey-white matter differentiation with expanded grey matter showing lower attenuation. The basal ganglia are not sharply defined. There is also generalized effacement of cerebral sulci.
Diagnostic workup
MRI with the apparent diffusion coefficient of water is becoming a sensitive tool of neuronal physiology and may represent a reliable indicator of progressive neuronal injury following cerebral ischemia.
White matter demyelization and necrosis
Sub cortical u fibers
Corpus calusum
Int/ext capsules
Gray matter
Neuroradiology, Grossman 353-4
Diagnostic workup
EEG can be a sensitive indicator of cerebral function.
the EEG can be used to assess cerebral function and to guide therapy.
Coma Patterns
Alpha Coma, Burst Suppression
Periodic spiking , Triphasic waves
Electrocerebral inactivity
Diagnostic workup
EEG has been used to assess prognosis. In patients with terminal coma, onset of abnormal EEG changes may sometimes be suggestive of a poor outcome :
(Rothstein et al 1991)
Burst suppression pattern
Periodic pattern( Generalized periodic discharges, periodic lateralized epileptiform discharges.)
Diagnostic workup
BURST SUPRESSION
Diagnostic workup
Periodic lateralizing epileptiform discharges may occur following focal cerebral insults such as infarction, but do not necessarily indicate active epileptic activity.
Diagnostic workup
Alpha coma is frequently associated with cardiac arrest, but its prognostic significance is unclear.
EEG pattern does not change with stimulation.
( ? Extremely poor prognosis PEDLEY,358)
Diagnostic workup
If seizures develop and aggressive management is desired for the patient, then epileptic activity should be treated to reduce further neuronal cell loss in the cortex. Status epilepticus, status myoclonus, and myoclonic statusepilepticus are associated with inability to recover consciousness in cardiac arrest survivors (Krumholz et al 1988)
ICU? – Consult ! – Prognosis?
Prognosis and complications
Prediction of outcome following coma has been the subject of multiple clinical studies
Earlier reports have noted that post-arrest coma longer than 3 days carried an unfavorable prognosis (Bell and Hodgson 1974)
Prognosis and complications
Several studies even report some permanent neurologic sequelae if coma duration is greater than 6 hours (Maiese et al 1988)
Others have correlated outcome with the pattern of motor responses and the presence or absence of particular brainstem reflexes.
( Levy et al , predicting outcome from hypoxemic-ischemic coma 1985)
Levy examined 210 patients in whom cardiopulmonary failure was the cause of coma and correlated clinical signs with outcome at 1 year .
Patient age, sex, location of the initial insult, etiology of the coma, and the presence of generalized seizures did not influence the degree of recovery.
Levy et al 1985
In contrast, the clinical examination did correlate with recovery. Individuals without pupillary light reflexes at the initial examination never regained independence. Absence of corneal reflexes following the first day was also a poor prognosis.
Levy et al 1985
Prognosis and complications
Increased numbers of brainstem reflex abnormalities were associated with reduced survival
Snyder et al 1977 ,Rudolf et al 2000
The most favorable sign of a good outcome
(can be rare at day 1):
Any form of speech, orienting spontaneous eye movements, intact oculocephalic or oculovestibular responses, ability to follow commands, and normal skeletal tone.
Prognosis and complications
Additional investigations seek to identify cellular markers that may signal a poor neurologic prognosis, such as elevated levels of S-100B and interleukin-8 ,(Mussack et al 2002)
Mussack et al 2002
Serum S-100B and interleukin-8 as predictive markers for comparative neurologic outcome analysis of patients after cardiac arrest and severe traumatic brain injury
Significantly elevated S-100B and interleukin-8 serum levels 12 hrs after cardiac arrest suggest that primary brain damage and systemic inflammatory response are comparably serious with that of traumatic brain injury. In both collectives, increased S-100B values measured 12 hrs after insult correlated well with an unfavorable neurologic outcome after 12 months.
Decorticate or flexor responses occur after damage to the hemispheres, or in cases of diffuse depression of cortical function following cerebral ischemia. Decerebrate or extensor responses correlate with destructive lesions of the midbrain and upper pons, but also may be present in anoxic encephalopathy. The absence of motor response, especially if flaccidity and areflexia are present, indicates severe brainstem depression and is frequently found in terminal coma or in severe sedative intoxication. Withdrawal and localizing responses imply purposeful or voluntary behavior. Obeying commands is the best response and marks the return of consciousness.
Neurology

Anti Epileptics
Na channel blockers:
Depakore- valproike acid: ( VPA )
Enz inhibitor Pro binding 95%
increase INR ! hypoglycemia
side effect fat , pco,hairy , pancreatitis , no rash !
increase ammonia level ,don't freak out if asymptomatic
asprine displace it from pro/ and increase its level.
Lamictal-lamotrigine ( LTG )
first line generalized sz
Rash
Pregnancy precaution
Keppra-levetiracetam ( LTC )
renal excretion
UMOA
Topomax-topiramate (TPM )
Enz inducer hperglycemia (metformin),decrease INR (Coumadin)
Diamox (used for NPH) like effect tingling and numbness
Dilantin-phenytoin ( PHT )
pro binding 90% increase INR ! hypoglycemia
Enz inducer decrease INR, hperglycemia
Half life 22h
Side effect : megaloblastic anemia,dupuytran contracture, rash
PHT decrease CBZ level to one third !!! , but CBZ increase dilanine level !!!
Tegretol-carbatrol-carbamazepine ( CBZ )
may induce absence status
Enz inducer decrease INR, hperglycemia
typical case: nursing home pt on this drug, physican prescrib darvocet or propoxiphen for pain and pt goes to coma (altered mental status) !!( increase level )
Side effect :hyponatremia , 10% rash ,diplopia
PHT decrease level to third !!
Cimetedine, verapamil,diltiazem, propoxyphen ,erythromycine isoniiazide increase the level.
trileptal-oxacarbamazepine
worse than carbamazepin in hyponatremia induction !
-------------------------------------------------------------------------------
CA channel blocker-only one !
zarontyn-ethosuximide absence sz
side effect : ataxia, rash
-------------------------------------------------------------------------------
GABAERGICS (most have ga, ba in their names ), (chloride chane GABA 1 , K increase and Ca decrease GABA2)
Primidone-Mysoline ( PRM ). Will metabolize to PB and other metabolites
Side effect : impotence,….
felbatol-felbamateside effect aplastic anemia and liver dysfunction
tiagan-tiagabine-gabatril
neurontin-gabapentinrenal excretion
Phenobarbital ( PB )hard to taper offEnz induce decrease INR, hyperglycemia
Side effect : dupuytran contracture , rash
Zonegram-zonisamide ( ZNS )diamox like effect tingling and numbnessSulfa allergy
Vigabatrinretinal deposition
which medication can normaliz the EEG:Depakote and PHB can normalize the EEG don't get full
case:if 21 year old pt with generalized sz new onset no etiology find, is geneticand need to be on med for the rest of life , don't full with sz free episodes or normal EEG
--------------------------------------------------------------------------
Therapeutic levels
Carbamazapin 5-10 ug/ml ,
Dilantin 10-20
Phenobarbital 20-40 , 90-120mg po phs, could start iv 300-800 mg iv. load for status epilepticus could use 10-20 mg/kg iv
Valporate 50-100, 10-60mg/kg TID (250cap),
-----------------------------------------------------------------------------
LOADING
Only : PHT/VPA/PB
Loading for pht and vpa the same 10-15 mg/kg
Expect for each gram the level raise 10
PB : don’t load more than 200 the first time (respiratory distress and ..), but you can reload 100 or 60 till level therapeutic later on /
H/F LIFE
Subscribe to:
Comments (Atom)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}